Conclusions and Future Problems
Application of multiplex fluorescent mRNA in situ hybridization
The significance of this project is not the introduction of new technology. Rather, its novel contribution is the effective combination of existing techniques to attain a robust and extensible protocol for the visualization of multiple mRNA expression patterns in single specimens. The advent of genome-scale analysis of gene expression in embryos, through transcriptional profiling with microarrays, compilation of databases of in situ expression patterns (e.g., the BDGP gene expression project, Tomancak et al., 2002), and bioinformatical modeling of cis-regulatory sequences (e.g., Stathopoulos et al., 2002), will stimulate many questions concerning the transcriptional regulation of large sets of genes. To address these questions, it will be necessary to make precise descriptions of the spatial and temporal relationships of gene expression patterns, demanding that they are viewed together in single embryos. Although in situ techniques will not soon match the breadth of microarray data, image registration methods supplemented with statistical classifications of large numbers of stained embryos might allow the strands of data provided by multiplex in situs to be woven together into accurate representations of the pattern of the embryonic transcriptome (see FlyEx, the Drosophila segmentation genes expression database, Myasnikova et al., 2001; Kozlov et al., 2002). It is now realistic to envision the construction of multidimensional gene expression datasets for any of the well-defined developmental modules that shape embryos, integrating the patterns of hundreds of genes at cellular resolution. Ultimately, both the quality of this form of gene expression 'metadata', and the effort required to perform the myriad experiments that support it, will be determined by the number of different mRNA targets that can be simultaneously detected in single embryos.
Closer to reality, monitoring the transcription of even a relatively small number (3-5) of genes together, with the superior resolution provided by fluorescence, can reveal subtleties in the relationships of patterns that are invisible with traditional histochemical methods. The study of gene regulation in development frequently encounters situations where a variety of transcriptional states exist on very fine scales, not only among different cells that are proximate in space, but also in the same cell, proximate in time. The high signal-to-noise ratio achieved in this protocol through the use of Alexa-fluor conjugates has improved substantially the resolution of images produced with standard fluorescence imaging apparatus, making it easy to observe the details of mRNA expression patterns at the level of single cells. Moreover, this improved sensitivity, when coupled with a linear detection method, greatly facilitates the quantitative comparisons (both among different embryos, and within single embryos) that furnish a glimpse of the dynamics of mRNA accumulation.
A heirarchy of detection strategies
As summarized in the following table, the techniques presented here fall into a heirarchy of sensitivity. Currently, 'sensitivity' is a subjective judgement of the brightness of the fluorescent stain achieved for a given gene expression pattern. These judgements have been made by eye with normal epifluorescence, and confirmed during confocal imaging with the use of look-up tables (LUTs) that color-code images according to pixel intensity.
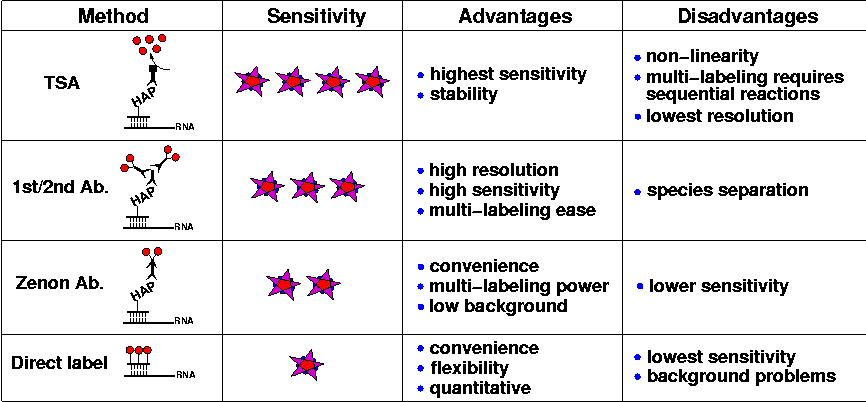
Three of these techniques use immunofluorescence to detect hybridized riboprobes labeled with a variety of haptens (HAP).
- Fluorescent tyramide signal amplification (TSA): The enzyme-catalyzed deposition of fluorophores near the site of probe hybridization produces an extremely bright fluorescent signal, and after extensive washing to remove the unbound tyramide substrate from embryos, the signal-to-noise ratio of the stain is excellent. Moreover, since the dye molecules are covalently bound to the tissue matrix of the embryo, the stain is stable over a period of months to years. The main drawback of TSA is that sequential reactions are required to detect more than one target. Other problems stem from the enzymatic nature of the stain: the small diffusion radius of the activated tyramide results in a loss of resolution; and the non-linear accumulation of fluorescent signal hinders meaningful quantitative comparisons. Nonetheless, TSA has become a very popular method due to its sensitivity and reliability.
- Anti-hapten primary antibody, fluorescent secondary antibody (1st/2nd Ab.): Employing primary antibodies to recognize haptenylated riboprobes, followed by secondary immunofluorescence, is probably the most attractive method explored here. We have found that secondary antibodies conjugated with the fluorescent dyes of the Alexa series (Molecular Probes) allow the detection of many different mRNAs expressed in a wide variety of cellular contexts and throughout development. With the appropriate primary antibodies and incubation conditions, bright fluorescent stains with low background are produced; and it is straightforward to label multiple expression patterns with the correct antibody cocktail. Also, the tight association of antibodies with their riboprobe targets enables high-resolution fluorescence imaging through which sub-cellular detail is easily percieved. The only difficulty with this technique arises from the potential for cross-species binding of the various antibody components.
- Anti-hapten antibody in fluorescent Zenon complex (Zenon Ab.): An alternative antibody-based probe detection method is the use of primary/secondary antibody complexes made with the Zenon labeling reagents (Molecular Probes). Although the fluorescent signals produced by these complexes are less bright than with traditional indirect immunofluorescence, background staining tends to be low, so that the signal-to-noise ratio of the resultant images is very good. The major benefits of this technique are the simplicity of performing a single post-hybridization incubation step, and the possibility of combining in the same experiment multiple primary antibodies raised in the same host species. For example, several high-affinity anti-hapten antibodies are mouse monoclonals and with this technique they can all be used together.
The fourth technique employs riboprobes into which fluorescent dye molecules have been incorporated.
- Directly labeled fluorescent riboprobes (Direct label): Direct label probes are very convenient to use, since they can be visualized almost immediately after hybridization, without intermediate incubation and washing steps. Also, since a single stock of aminoallyl-RNA can be covalently linked with any fluorescent dye, direct labeling provides flexibility in the design of multiplex experiments. Moreover, since the fluorescence of these probes can be calibrated, it is possible to make direct, in situ, quantitative measurements of mRNA metabolism (Femino et al., 1998). But the great promise of this method is that the potential number of specific mRNA targets that could be detected in single specimens becomes the same as the number of spectrally resolvable fluorophores (see below). Despite the success of directly labeled DNA probes in chromosome M-FISH, and also for mRNA detection in tissue culture cells (Levsky et al., 2002), we have had difficulty attaining high-quality fluorescent labeling of embryos with Alexa-fluor substituted riboprobes. Using these probes, we have produced acceptable images only of genes that either are expressed at very high levels or (due to unknown factors influencing the efficacy of in situ hybridization) always can be detected well, even under dubious experimental conditions! Although we have not yet empirically explored the possible causes of the problem, it seems likely that we are confronting a severe loss in hybridization efficiency due to the high substitution rate of Alexa dye molecules in these probes; and perhaps particular Alexa-fluors are more crippling than others. Also, these probes exhibit a significant non-specific binding to embryos under standard hybridization and washing conditions, leading to speckled background fluorescence.
In conclusion, the choice between these approaches to detecting mRNA with fluorescence should be made according to the challenges defined by particular experiments. How many different mRNAs do you want to detect? How abundant is the transcript, or how difficult was it to detect in previous experiments? Are you interested in viewing broad domains of expression, or sub-cellular localization of transcripts? Do you want to make quantitative comparisons between embryos? Any of these four methods can be combined in a specific protocol according to the benefits provided by each.
How many expression patterns?
What are the technical issues that limit the number of expression patterns that can be detected together in single embryos? Will it be possible to visualize 10-20 different genes? Nascent transcript M-FISH (discussed here under Nuclear Dots and Virtual Colors) will provide one approach to this problem; alternatively, how far can the useful repertoire of haptens, antibodies, and fluorescent dyes expand? Consider this diagram, which schematically represents the methods presented here:
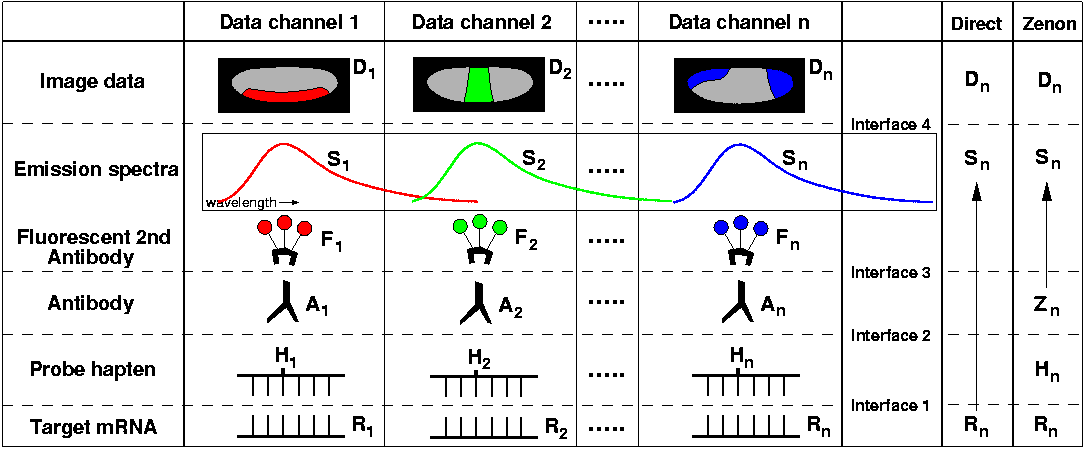
Probes that hybridize specifically to each target mRNA (R1-Rn) are labeled with different haptens (H1-Hn), which in turn are recognized by specific antibodies (A1-An) raised in immunologically distinct host species. Secondary antibodies conjugated with different fluorochromes (F1-Fn) are applied, and from the resolvable emission spectra (S1-Sn) of these fluorochromes, separate image data (D1-Dn) is collected for each expression pattern. In essence, a unique 'data channel' is established for each gene, linking transcripts immobilized in the embryo to pixel values arrayed in an image file. As data channels are assembled, the 'interface' between each added element must be highly specific in order to maintain separation between channels. The requirement for specificity at each interface imposes a variety of constraints on increasing the number of gene expresssion patterns visualized with this kind of protocol. Below, issues associated with each interface are discussed in turn:
- Nucleic acid hybridization: If the sequences of cohybridized riboprobes are sufficiently unique, cross-hybridization will be minimized, and it seems reasonable to suppose that a very large number of different probes could be hybridized to embryos before encountering problems. Thus far, we have successfully hybridized up to seven different riboprobes derived from cDNAs. The greater difficulty is not the specific hybridization of probes to mRNA targets, but the specific detection and visualization of those hybridized probes; in this model of data channels, direct label probes have the advantage of skipping over these intervening steps, proceeding immediately to the stage of spectral separation of fluorescence (diagram, second column from right).
- Hapten substitution and antibody recognition: We have attempted to use only four different haptens in this protocol (DIG, BIO, FITC, and DNP), but other hapten-modified nucleotides, and their antibody partners, are commercially available. For example, one M-FISH method employed tetramethylrhodamine (TMR)-modified nucleotides in addition to the four just mentioned, detected with rabbit anti-rhodamine antibody (Henegariu et al., 2000). Other fluorescent dyes can be incorporated into riboprobes and used as haptens, including Texas Red, Alexa Fluor 488, BODIPY FL, and Cascade Blue. Another possibility is estradiol-modified nucleotides, offered by Roche for a short time but then discontinued. The lack of a wider array of haptens would constrain this protocol to detecting no more than 6-8 mRNA targets, although perhaps in the future many more modified-nucleotide/antibody pairs could be designed.
- Species separation of secondary antibody binding: To preserve specificity across the two antibody layers, each anti-hapten antibody must be from a different host species. Even so, this array of host species must be carefully selected to avoid cross-species interactions. For example, using anti-goat and anti-sheep secondaries together resulted in unacceptable cross-talk between image channels. Also, since the majority of commercially available anti-hapten antibodies are from a relatively narrow range of host species (rabbit, mouse, goat, and sheep), this step presents a serious impediment to increasing the number of available data channels. However, Zenon antibody labeling removes these difficulties ('Zn', in the far right column of the diagram), and also it should be possible to create primary antibodies in additional compatible species, such as guinea pig, chicken, dog, or human (I'll volunteer, but can't guarantee a terminal bleed).
- Spectral separation of fluorescence: Ultimately, all multiplex fluorescent techniques rely on the ability of the detection apparatus to separate the light emitted by the different fluorescent dyes which label the specimen. Unfortunately, the emission spectra of most fluors are wide, so that when multiple fluors are used together, their emission peaks must be spaced apart; and since the total spectrum space is limited, so is the total number of fluors that can be observed at once. However, innovations in microscopy hardware and software alleviate the traditional problem of spectral overlap. First, mathematical analysis of the spectrum of emitted fluorescent light, termed 'linear unmixing', has been implemented on the latest generation of confocal microscopes from Zeiss (LSM510-META), allowing the separation of signals from dyes with overlapping emission spectra (Dickinson et al., 2001). Even more powerful is a form of microscopy, applied in the cytogenetic FISH technique 'spectral karyotyping' (SKY), that is capable of separating signals from fluors with almost identical emission spectra, using interferometry coupled with Fourier analysis (Schrock et al., 1996). This technology has been applied also to imaging seven different fluors in immunolabeled tissue sections (Tsurui et al., 2000). Other advances in fluorescent microscopy instrumentation, such as the 'filter-free' confocal system from Leica (SP2-AOBS), provide flexibility in the configuration of excitation laser lines and emission collection wavelength ranges, making it easier to separate the signals from a greater number of fluors.
The question remains, how many expression patterns? The answer depends on the availibility of new reagents, and the progressive developments in fluorescence imaging technologies. By trying out some of the possibilities mentioned above, with any luck, we will soon be able to visualize six expression patterns per embryo, and eight does not seem beyond imagination.
Previous: Nuclear Dots and Virtual Colors
Back to Index