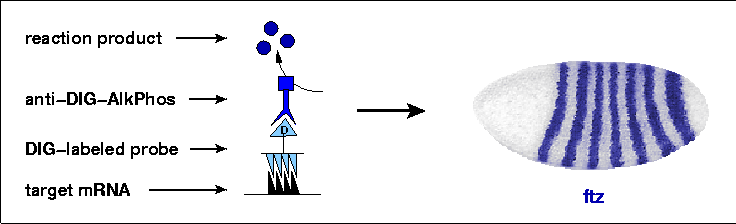
After hybridization, probes are recognized by an anti-digoxigenin antibody conjugated with alkaline phosphatase. During incubation with the substrate NBT/BCIP, this enzyme deposits a brownish purple precipitate in the embryos, which then turns a deep blue color after exposure to ethanol. In this way, cells which have produced a particular mRNA are stained, and the pattern of stained cells across an entire embryo can be seen, such as the simple seven-stripe pattern of the ftz gene shown here. This method has been applied successfully to embryos of many species, and is now being put to use on a massive scale by the Berkeley Drosophila Genome Project (BDGP) to determine the embryonic expression patterns of thousands of genes. The initial results of this effort have been published (Tomancak et al., 2002) and these annotated expression patterns can be viewed in this in situ database curated at the BDGP.
Soon after the introduction of this method, it was extended so that the expression patterns of more than one gene could be seen in single specimens. In one approach, both biotin- and digoxigenin-labeled probes are hybridized to embryos, and signals from each are enzymatically developed with different color reaction products. After the detection of the digoxigenin probe as described, the biotin probe is detected with antibodies conjugated with horseradish peroxidase, which deposits a reddish brown reaction product when exposed to the substrate DAB (O'Neill and Bier, 1994). With this method, contrasting blue and brown stains make it possible to distinguish two different expression patterns, and thus to describe their spatial relationship. Another approach, using both fluorescein- and digoxigenin-labeled probes, achieves two color staining by performing two sequential alkaline phosphatase reactions: the first using the standard NBT/BCIP substrate; and the second using Fast Red, which results in a bright red reaction product (Kosman and Small, 1997). Extending these methods even further, three expression patterns can be detected with fluorescein, digoxigenin, and biotin probes and three sequential alkaline phosphatase reactions (Hauptmann, 2001).
A major limitation of these histochemical multi-labeling techniques is that overlapping regions of two expression patterns are difficult to discern. In cells where both reaction products are present, one product may obscure the presence of the other, depending on their relative abundance and color. The general problem is how to separate signals from molecule-specific probes binding to different targets in the same location. The solution is to tag those probes with fluorescent dyes which emit light in defined wavelength ranges, as demonstrated here for triple fluorescent staining of proteins:
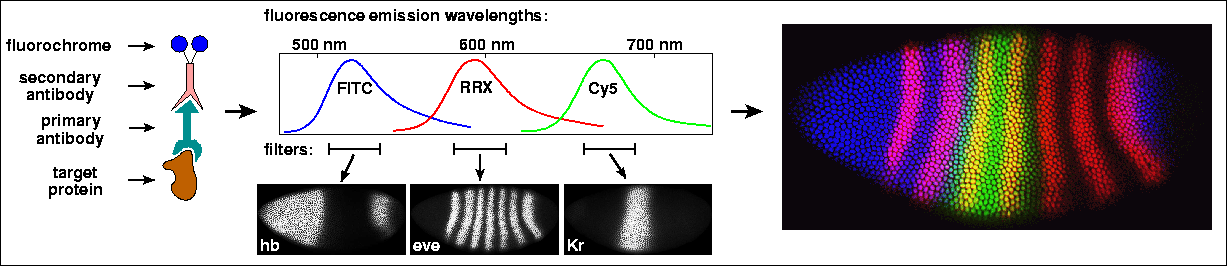
Fluorescent labeling of proteins in Drosophila embryos was already commonplace at the time when the whole-mount immunohistochemical mRNA in situ method was introduced. As shown in the example above, three fluorochrome-tagged secondary antibodies label three primary antibodies, which in turn recognize three transcription factor proteins, hb, Kr, and eve (left). The fluorochromes indodicarbocyanine (Cy5), rhodamine red-X (RRX), and fluorescein (FITC), emit light in different parts of the spectrum, so that three separate images of the embryo can be collected with the appropriate color filters (middle). These images, each representing the expression pattern of a single protein, can be color-coded and merged, so that the spatial relationships between the patterns are easily perceived (right). In particular, overlapping patterns are distinguished by mixtures of colors: in the example above, where the red stripes of eve overlap with the green band of Kr, they appear yellow; and where they overlap the blue cone of hb, they appear magenta (Paddock, 2001).
Fluorescence techniques were then adapted to whole-mount in situ hybridization protocols. First, since the reaction product of Fast Red produces strong red fluorescence (in addition to being visible, as mentioned above, with bright-field illumination), using it in place of the traditional NBT/BCIP substrate enabled fluorescent detection of mRNA (Kagiyama et al., 1993). Double fluorescent labeling was achieved with sequential alkaline phosphatase reactions: the first with Fast Red, and the second producing the bright green-yellow precipitate of the ELF97 (enzyme-labeled fluorescence) substrate (Jowett, 2001). An important advance was the application of fluorescent secondary antibodies to the detection of hybridized riboprobes, similar to the fluorescent detection of proteins outlined above (Hughes et al., 1996). Not only did this approach enhance the spatial resolution of the RNA stains, but also set the stage for fluorescent multi-labeling without laborious sequential immunochemistry steps. With this technique, fluorescein and digoxigenin probes were visualized using primary antibodies specific to the probe labels and secondary antibodies conjugated with the bright green and red fluorescent cyanine dyes, Cy2 and Cy3 (Hughes and Krause, 1998). However, since non-enzymatic immunofluorescence methods are generally less sensitive, and since there are many fewer target mRNA than protein molecules in cells, enzymatic amplification of RNA signals is often beneficial, if not necessary. The use of fluorescent tyramide substrates in sequential horseradish peroxidase reactions (tyramide signal amplification, TSA) achieves double fluorescent labeling of mRNA with high sensitivity, but with higher resolution than the more diffusible reaction products of alkaline phosphatase (Wilkie and Davis, 1998).
A notable adaptation of the word 'multiplex' to the subject of biological macromolecules occurred with the introduction of a powerful karyotyping technique for human cytogenetics. 'Multiplex-fluorescence in situ hybridization' (M-FISH) assigns a spectral signature to each chromosome through specific combinations of different fluorochromes (Speicher et al., 1996). The concept behind M-FISH resembles the situation described above for the color mixtures resulting from fluorescent antibody labeling, and is more thoroughly explained here, under Nuclear Dots and Virtual Colors. Recently, 'multiplex' has come into more general usage in biology, implying the simultaneous identification of multiple molecular components of various preparations. This usage is close to its original sense in the world of electronics, where 'multiplex' conveys the idea of simultaneously transmitting multiple signals or messages through the same circuit. Large systems in which continuous streams of electronic data must be communicated, such as the space shuttle or the internet, rely on 'multiplexers' and 'demultiplexers' to send and recieve multiple messages over a single wire without mixing them up. In the context of detecting gene expression through in situ hybridization, one meaning of 'multiplex' could be drawn by the following analogy. A fixed embryo is like a single wire, and the expression patterns of all of the thousands of genes in that embryo are like many separate messages being transmitted. All of those messages are available to be read, or 'demultiplexed', off that embryo, but it is the choice of gene-specific probes that determines which of them actually are. Gene expression patterns are the messages; modified nucleotides, antibodies and fluorescent dyes are the devices that allow those messages to be independently received and recorded; and the goal of multiplex fluorescent mRNA in situ hybridization is to record as many of those messages as possible.
'Multiplex fluorescent mRNA in situ hybridization' is thus the sum of methods by which the mRNA of multiple genes can be visualized with fluorescence in single specimens. The protocol presented here is an adaptation of a whole-mount in situ protocol used in the laboratories of Mike Levine (Jiang et al., 1991) and Steve Small (Kosman and Small, 1997). It incorporates fluorescent detection strategies that are based on protocols developed by Hughes and Krause (1998), and Wilkie and Davis (1998), mentioned above. By combining and extending these strategies, four expression patterns per embryo can be visualized, and this number probably will soon increase. For some thoughts on the motivation for developing this capability, please see Conclusions and Future Problems.